
For decades, the standard script for testing generic drugs was nearly identical. Researchers would gather a group of young, healthy men. They would take two pills-one original brand, one copycat-and scientists would measure how much medicine ended up in their blood. If the numbers matched, the drug was approved for everyone. The logic was sound in theory: if the body absorbs the drug, does it matter who takes it? In practice, it mattered a lot.
This approach left out roughly half the human population-women-and ignored another huge demographic-the elderly. When medications designed for post-menopausal women or seniors were tested exclusively on 25-year-old males, we risked approving drugs that worked differently in real-world patients. As of 2024, agencies like the U.S. Food and Drug Administration, often referred to as the FDA, have shifted the goalposts. Now, Bioequivalence studies require careful balancing of age and sex to ensure safety across diverse groups.
What Does Bioequivalence Actually Mean?
Before diving into who gets tested, you need to understand what is being measured. Bioequivalence isn't about whether a pill cures your headache; it's about how fast and how much of that drug enters your bloodstream. Think of it like comparing two brands of coffee. One might taste better, but do they wake you up at the same speed? In pharmaceutical terms, this comparison happens between a reference product, typically the brand-name version and a generic alternative.
If the rate and extent of absorption are similar without significant differences, the products are considered bioequivalent. For years, the scientific community assumed that a difference in absorption rates seen in young men applied universally to everyone else. We thought that because humans share basic biology, a study in men translated perfectly to women, children, or seniors. Modern pharmacokinetics tells us this assumption was wrong. Gender hormones, body composition, and aging enzymes change how we process medication.
The Evolution of Study Populations
Historically, bioequivalence trials favored convenience over representation. Using young, healthy males minimized variables. Their hormonal cycles were stable, their metabolism consistent, and there was less risk regarding pregnancy complications. But this created a "homogeneity bias." Drugs specifically intended for women, such as levothyroxine for thyroid issues, were often tested on men because finding female volunteers was harder or costlier.
By the early 2020s, the science caught up with the social reality. We realized that Pharmacokinetics involves distinct metabolic pathways that differ significantly between sexes. For example, certain enzymes metabolize drugs slower in females due to lower levels of CYP2D6 activity in some contexts. If a drug relies on those enzymes, a dose safe for a male might be toxic for a female. Ignoring this leads to adverse reactions once the drug hits the pharmacy shelves. Consequently, regulatory bodies moved away from "healthy volunteer" defaults toward "representative population" models.
Current Regulatory Standards
The rules governing who participates in these studies vary by region, though they are converging. The FDA's 2023 draft guidance was a watershed moment. It explicitly states that if a drug is meant for both sexes, the study should include similar proportions of males and females. Specifically, they recommend a ratio close to 50:50 unless there is a scientific reason not to.
| Agency | Age Requirements | Sex Representation |
|---|---|---|
| FDA (USA) | 18+ (60+ required for geriatric drugs) | Balanced (~50% each) |
| EMA (Europe) | 18+ preferred | Either sex (no strict mandate) |
| ANVISA (Brazil) | Strictly 18-50 | Equal distribution |
In contrast, the European Medicines Agency, commonly known as the EMA, maintains slightly different guidelines. Their 2010 guideline emphasizes sensitivity over representativeness. They state subjects can be either sex, but they prioritize detecting formulation differences. This creates a gray area where sponsors might stick to the "healthy male" default if they aren't challenged during review. However, ANVISA in Brazil takes the opposite extreme, limiting age strictly to 18-50 to avoid variability, which ironically excludes the very elderly patients who often rely on generics the most.
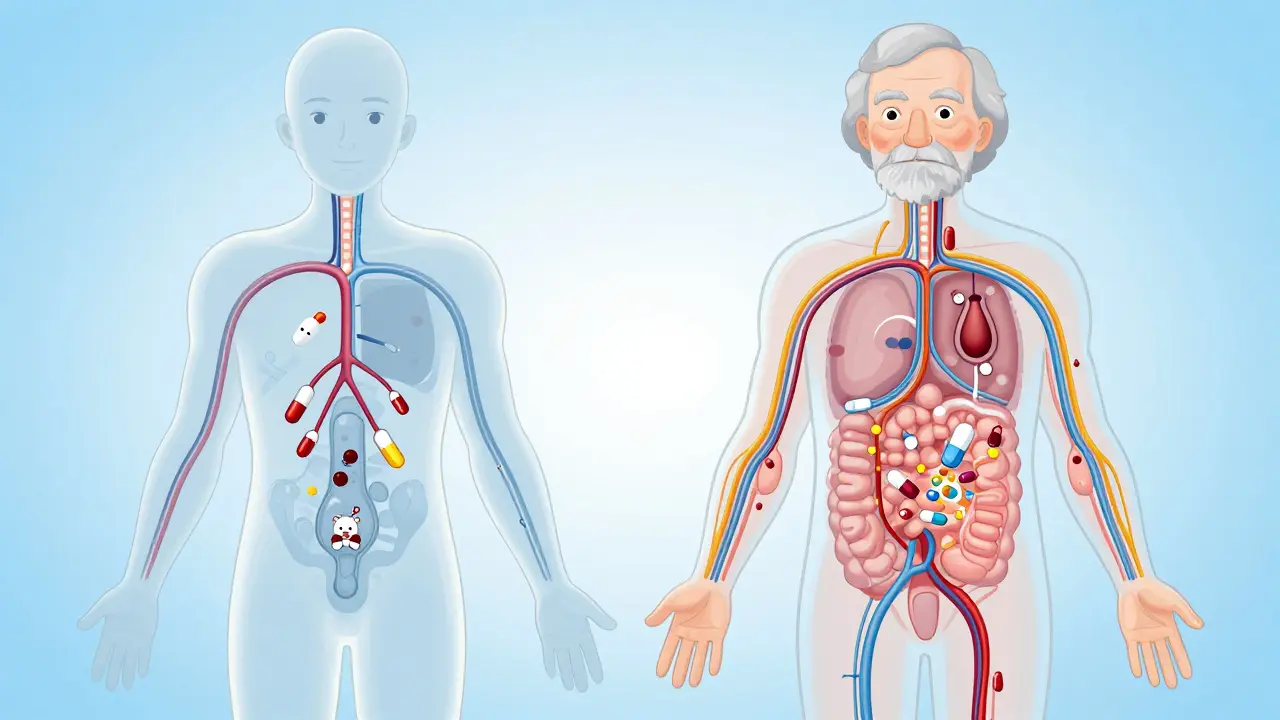
Practical Challenges in Recruitment
Writing the policy is easy; executing it is messy. Achieving a balanced gender split costs more money. Recruiters report that women participate in clinical trials at lower rates due to family responsibilities and scheduling conflicts. Sponsors face a 20-30% increase in recruitment costs when targeting equal ratios. Additionally, excluding women from studies of drugs meant for women creates a paradox. A trial for contraception cannot ethically exclude women, yet historically, many hormonal trials used male participants to bypass cycle tracking complexities.
Statistical power also plays a role. Smaller studies, those with fewer than 24 participants, are prone to "false signals." An outlier result from one person might look like a gender difference when it's just random noise. Larger studies (36+ participants) smooth out these anomalies. This means smaller companies developing niche drugs might struggle to afford the sample size needed to prove bioequivalence fairly across genders.
Why Elderly Inclusion Matters
While gender parity has been the headline, age remains a critical factor. The kidney function declines naturally with age, affecting how long a drug stays in the body. Younger adults often clear medicines faster. If we test a new heart medication only on 25-year-olds, we might approve a dosage that builds up to toxic levels in an 80-year-old. The FDA now allows subjects aged 60 and older in studies, acknowledging that renal impairment is common in the real world.
However, including seniors introduces its own hurdles. Older volunteers often have comorbidities (multiple health conditions) and take other medications. To maintain scientific purity, researchers prefer people who take nothing else but the study drug. Balancing the need for "pure" data against the need for "real-world" safety is the tightrope modern researchers walk. The compromise often involves stable patients with chronic conditions rather than purely "healthy" seniors.

Future of Diverse Testing
We are moving toward a model where "special population" is no longer a footnote but a standard requirement. By 2025, major agencies expect full justification for any single-sex enrollment. This pushes the industry to develop better statistical methods to analyze sex-based interactions. There is talk of developing specific criteria for Narrow Therapeutic Index drugs, where tiny differences in blood concentration can mean the difference between healing and harm.
Ultimately, the shift improves public trust. When patients see that drug approval involved people who look like them, confidence in generics rises. We are transitioning from a "safe enough for the average man" standard to a "safe for everyone" standard. While the transition period brings higher costs and longer timelines, the reduction in adverse events among women and seniors justifies the investment.
Frequently Asked Questions
Why were women excluded from historical bioequivalence studies?
Historically, researchers excluded women to minimize variability caused by hormonal cycles and to avoid pregnancy risks during testing. They sought the most homogeneous group possible, which turned out to be young, healthy males, leading to data gaps.
Does the FDA require equal gender representation in studies?
Yes, according to the 2023 draft guidance, applicants should include similar proportions of males and females (roughly 50/50) for drugs intended for both sexes unless a scientific justification exists for deviation.
Can bioequivalence results from young adults apply to seniors?
Not automatically. Because aging affects metabolism and kidney function, extrapolating results requires justification. Specific drugs targeting the elderly often mandate including subjects aged 60 or older in the trials.
How do cost factors influence study design choices?
Balanced recruitment increases costs by 20-30% due to longer timelines and lower participation rates among women. This sometimes discourages sponsors, though regulatory pressure is shifting acceptance toward inclusive designs.
Are there different rules for European vs US approvals?
Yes. The FDA explicitly mandates balance, whereas the EMA prioritizes study sensitivity over representativeness. However, global convergence suggests the EU may adopt stricter diversity requirements in future updates.
I think the shift towards balanced recruitment is finally acknowledging how messy real human biology actually is.
We spent so long trying to remove variables that we removed the actual patients who need the drugs.
It feels like we are correcting a massive blind spot in how pharmaceutical development works.
Hopefully this encourages sponsors to stop viewing diversity as an obstacle to clear.
This is such an important point to bring up right now 😊
The fact that women participate less due to family responsibilities adds another layer to the problem 💔
We really need better support systems for volunteers in these trials 🙏
Thank you for highlighting the cost factor so clearly 👍
The financial implications cited regarding the twenty percent increase in costs are often understated in these discussions.
Pharmaceutical budgets operate on razor thin margins for generics and any additional variance eats profitability.
Recruiters struggle significantly when targeting equal gender ratios across all therapeutic areas.
Regulatory pressure forces compliance but ignores the logistical reality of scheduling conflicts.
Companies will find loopholes in the guidance unless enforcement becomes strictly binary.
You completely miss the ethical imperative behind these mandatory changes!!!
Safety data cannot prioritize profit margins over patient outcomes!!!!
The long term liability of adverse reactions dwarfs the initial recruitment costs!!!!!
Regulators are acting correctly by demanding representation of the actual patient pool!!!!!
Every delay in approval saves lives in the long run!!!!!
From a pharmacokinetic standpoint the distinction in metabolic pathways is critical.
CYP2D6 activity variations between sexes create significant divergence in drug exposure profiles.
If the area under the curve differs beyond the eighty to one hundred twenty five percent bounds the product fails.
Generic developers must now account for sex based clearance rates in their power calculations.
Bioanalytical methods need higher sensitivity to detect these nuanced deviations in steady state concentrations.
Your reliance on enzyme kinetics suggests a fundamental lack of understanding regarding clinical trial design.
These minor enzymatic shifts rarely translate to statistically significant clinical outcomes in generic equivalence.
The industry standard has evolved precisely because such granular analysis proves inefficient for most formulations.
Only truly sophisticated practitioners understand that formulation differences dominate over minor metabolic variances.
Most of this regulatory guidance is performative compliance rather than scientific necessity.
Excluding women from contraception trials was the single biggest failure in modern medicine history!
The comparison between agencies like the FDA and EMA highlights interesting regional cultural differences in science policy.
In India we often see stricter adherence to age limits to preserve data purity compared to western markets.
It is important to recognize that one size does not fit all when defining representative populations globally.
Harmonization of these standards would greatly benefit multinational drug development timelines.
We should appreciate the progress while respecting local regulatory contexts.
Seeing the trend move towards full justification for single sex enrollment gives me a lot of hope for the future.
By twenty twenty five we should see a much clearer picture of safe dosing for elderly patients.
Public trust in generics will definitely rise once people know their specific demographics were tested.
It takes a village to get safety right and this shift includes everyone involved in the process.
Keep pushing for transparency and the industry will continue to improve its practices.
The fundamental issue lies in our historical prioritization of convenience over safety.
For too long we have accepted data gaps because they were easier to manage.
Young men were simply available subjects who did not complicate the statistical models.
Now we see that biology does not respect those convenient boundaries.
Hormonal fluctuations dictate absorption rates in ways that static tests cannot capture.
Kidney decline in seniors creates toxicity risks that remain invisible in healthy cohorts.
We are finally paying the price for decades of homogenized trial designs.
The cost increase mentioned is actually an investment in public trust rather than a budget burden.
Safety margins must expand when the population expands.
It is unethical to approve medication based on half the demographic spectrum.
Regulatory bodies are slowly waking up to this glaring oversight.
Brazil and Europe show different paths toward the same necessary destination.
We must question every exclusion criterion applied to special populations.
Science demands representation just as much as it demands accuracy.
Future generations should inherit a medical system that sees everyone as valid.